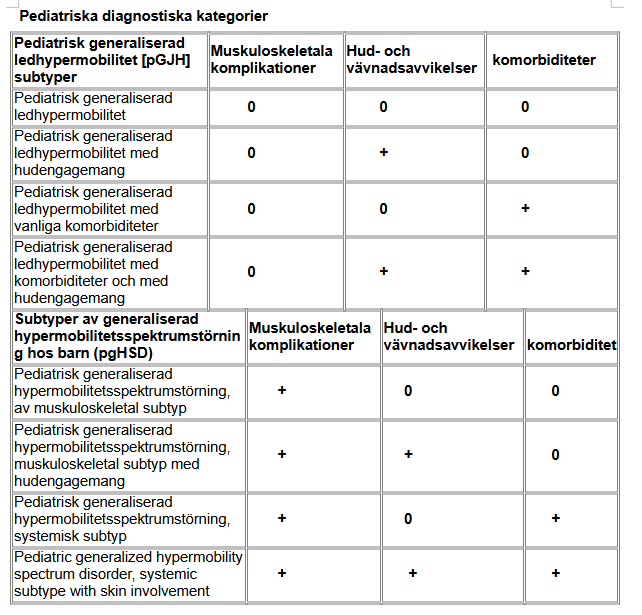
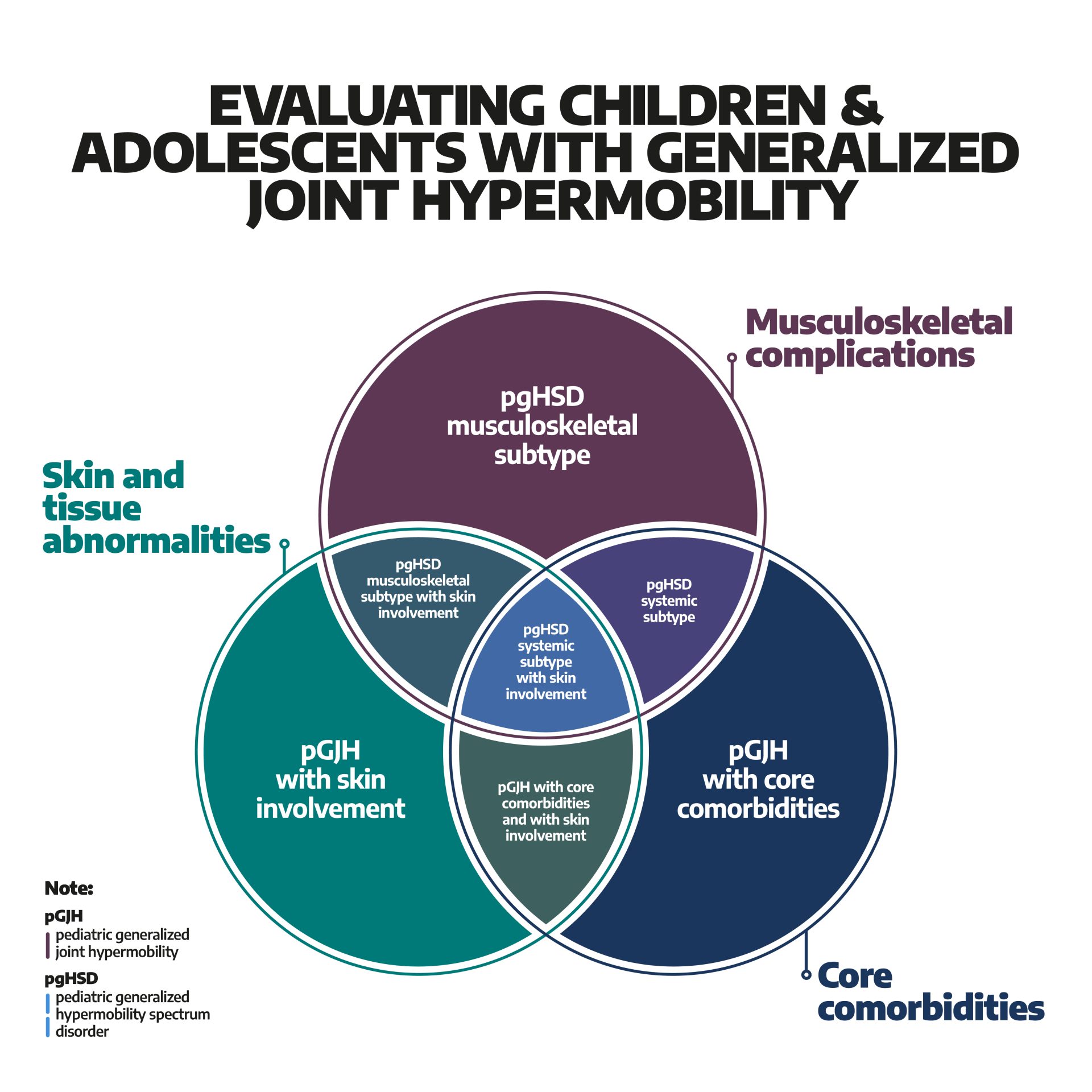
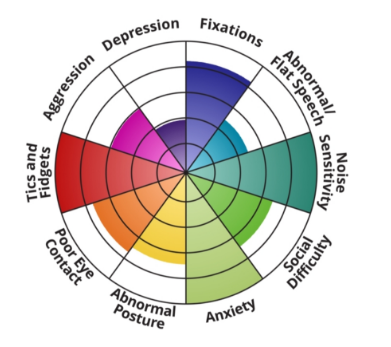
Anlag för hEDS och HSD i generna kan ställa till problem på många olika sätt. I värsta fall kan det bidra till att föräldrar bli anklagade för barnmisshandel och därmed förlora vårdnaden om barnet och t o m bli frihetsberövad.
Det finns två situationer där detta kan bli verklighet.
Jag tillämpar här begreppet hEDS/HSD för att täcka in hela det relevanta kliniska tillståndet.
1. Frakturer som upptäcks på späda barn då föräldrarna tagit kontakt med sjukvården p g a oro för sitt barn, som visat tecken på allmänt missnöje. Ofta finns inget känt trauma med i bilden ej heller synliga yttre tecken på våld. Men på röntgenbilder kan det förekomma diskreta skelettförändringar av en typ som har förknippas med barnmisshandel. Idag vet vi att dessa förändringar lika väl kan bero på benskörhet och uppstå vid normal hantering men också följd av en svår förlossning.
Den vanligaste orsaken till benskörhet är brist på vitamin D, d v s barnet har rakit, något som inte alls är särskilt ovanligt hos prematura eller underviktiga barn men också vid nutritionsrubbningar av olika slag hos modern. Även genetiska faktorer kan bidra till benskörhet varav Ehlers-Danlos anses vara ett exempel.
Denna benskörhet går under begreppet ”Metabolic bone disease” som beskrivs utförligt i en stor aktuell svensk studie
Orsaken till att man länge har förbisett brist på D-vitamin i dessa fall är att det måste analyseras direkt efter födseln. Man ger ju extra D-vitaminer till nyfödda barn vilket snabbt normaliserar blodhalten medan skelettförändringar d v s rakiten tar längre tid att läka. Genom att mäta D-vitamin i det s k PKU testprovet som sparas på alla nyfödda barn, kan låga halter vid födseln numera bekräftas i ett senare skede. Rakit är en röntgenologisk diagnos som kräver särskild kompetens och erfarenhet att bedöma. Sådan kompetens saknas i Sverige. Detta missförhållande har lett till att ett flertal familjer förlorat vårdnaden om sitt barn i vårt land(se boken Stulna liv)
Nu finns det således starka indicier på att benskörhet i nyföddhets-perioden förekommer under vissa betingelser och kan förklara suspekta röntgenförändringar utan att misshandel behöver misstänkas.
I ett aktuellt fall tvingades dock föräldrarna själva att bekosta expert-vittnen från utlandet (dyrt!!), vilket dock bidrog till frikännande och hemtagning av barnet.
Barnskyddsteamen har en viktig funktion att fylla men måste vara öppna för alternativa förklaringar och nya vetenskapliga rön.
2.Barnmisshandel kan också vara av mer psykisk karaktär och då avser jag begreppet Münchhausen by Proxy MbP se läkartidningen
Det handlar om svårtolkade oroväckande symtom hos barn som leder till upprepade kontakter med sjukvården utan att man hittar något fel på barnet eller annan vettig förklaring. Man upplever moderns oro obefogad och misstänker till slut att problematiken bottnar i ett patologisk behov hos modern att utsätta sitt barn för onödiga och obehagliga undersökningar. Moderns psykiska hälsa ifrågasätts i dessa fall och motiverar en orosanmälan.
Barnskyddsteamen är ofta snabba på att bekräfta tolkningen MbP d v s en form av barnmisshandel vilket brukar leda till att mor och barn skiljs åt genom tvångsåtgärder. Detta må vara helt korrekt under förutsättning att man har övervägt och uteslutit hEDS/HSD hos barnet.
Men idag saknas ofta kunskap om innebörden av hEDS/HSD och då framför allt den samsjuklighet som präglar dessa tillstånd. Dysautonomi är mycket vanligt, likaså uttryck för mastcells aktivering. Dessa avvikelser kan framkalla symtom som inte är ovanliga i befolkningen rent allmänt men är särskilt vanliga och framträdande hos barn och ungdomar med hEDS/HSD varav flertalet saknar fastställd diagnos, liksom även hos de anklagade mammorna i flertalet fall.
Därför krävs det goda kunskaper om hEDS/HSD när en medicinsk bedömning görs i dessa fall, särskilt när symtombilden är svårtolkad och ospecifik.
Ofta tolkar man ätproblem hos unga flickor som uttryck för ”Anorexia Nervosa” när det i stället är säljningssvårigheter p g a dysautonomi eller ospecifik överkänslighet p g a Mastcells aktivering.
Se bifogade artiklar nedan där man ibland nöjer sig med att misstänka hEDS/HSD utifrån generaliserad överrörlighet d v s GJH vilket ändå ger en antydan om bakomliggande sårbarhet.
Dessa horribla exempel på konsekvenser av vad hEDS/HSD kan ställa till med, belyser hur viktigt är att kunskap kring hEDS/HSD väl förankras inom sjukvården, inte bara i form av specialistmottagningar, utan kanske främst i primärvården, för det är där dessa patienter ofta hamnar först.
https://pubmed.ncbi.nlm.nih.gov/26506923/
https://pubmed.ncbi.nlm.nih.gov/35095619/